| 2_5 Female X Chromosome Informative SNPs (Version 1) | mcmillan / Version 34 |
This is the first version of Informative SNPs, which has been mentioned in May 24th meeting.
The tarball can be downloaded from here. Notice that the data set includes eSNPs and intergenic SNPs.
! A new version, Version 2, is available here.
Summary of the Informative SNPs dataset.(Figures only show eSNPs)
|
FG/GF
|
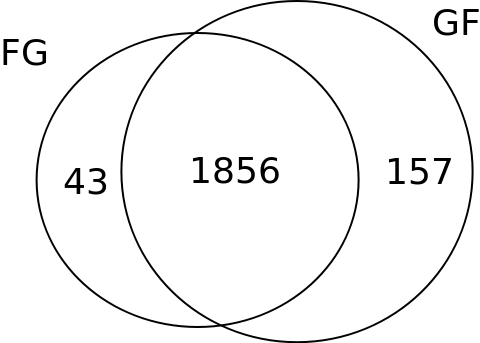 |
||||||||||||||||
|
FH/HF
|
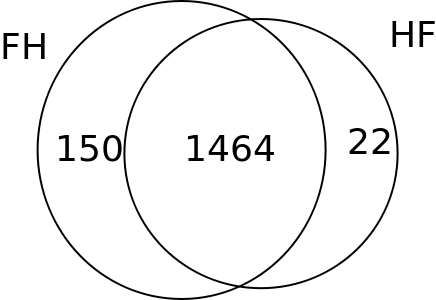 |
||||||||||||||||
|
GH/HG
|
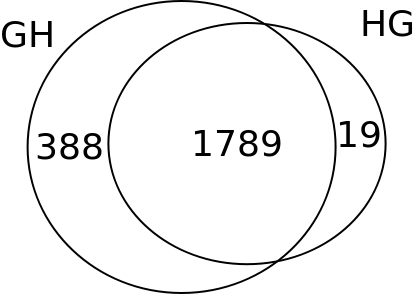 |
Detailed Procedure:
(0) For each F1 cross, we only consider the Sanger SNPs between the maternal and the paternal inbred strains. For example, for FG cross, we only consider SNPs between CAST and PWK.
(1) We first use the F1 samples, and compute at each SNP position the ratio of the sum of maternal allele and paternal allele to the pileup height. We remove loci where this ratio is less than 25% in more than 25% samples. (To ensure each variant is mainly caused by maternal allele or paternal allele but not by random noise)
(2) Then we consider the inbred strains and perform a similar procedure on both maternal and paternal inbred samples. However, the ratio and the criterion we used is different from the one in F1 cross. For maternal samples, we use the ratio of paternal allele to the pileup height and remove SNPs where more than 25% samples have the ratio greater than 25%. For paternal samples, we do it the other way around. (To ensure in maternal inbred strain each variant is mainly caused by maternal allele, and so as paternal inbred strain)
(3) The final output will be SNP positions either in Set1 and Set2 or in Set1 and Set3. Of course, SNPs that are in Set1, Set2, and Set3 are included. We will also remove SNP positions where less than 75% of samples have pileup height greater than 50. (To ensure each SNP is covered by enough pileup) This criterion is changed in Version 2.
In short, for FG cross, we remove the bad SNPs with the FG samples data, and we did the same procedure on its reciprocal cross GF. A SNP can be good in FG samples but bad in GF samples.